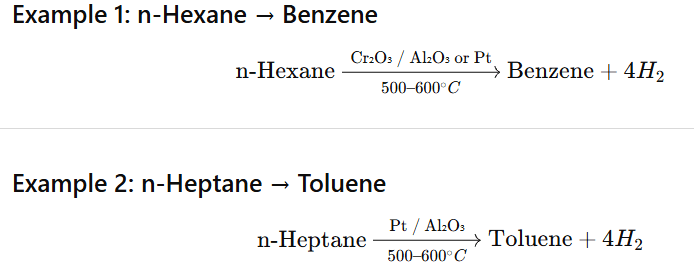
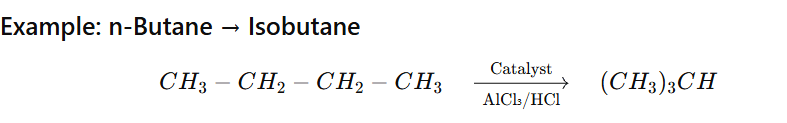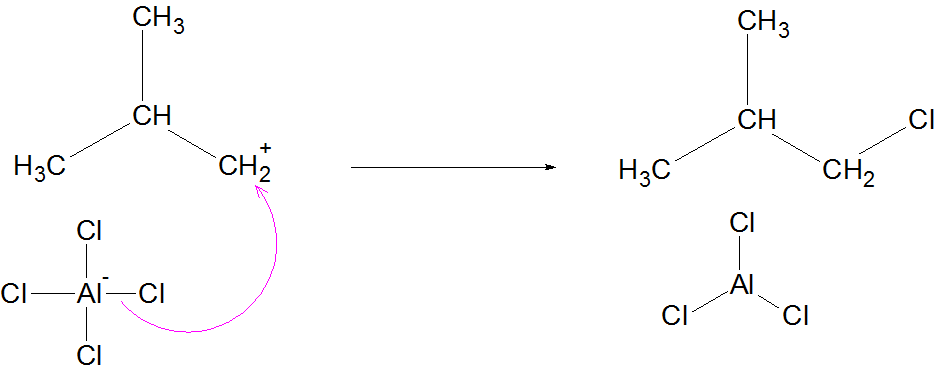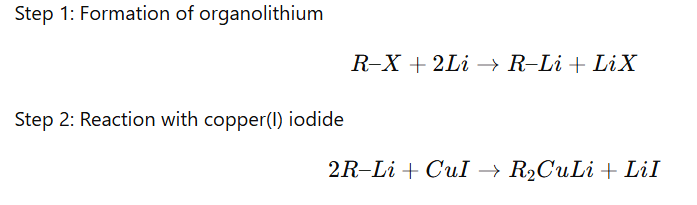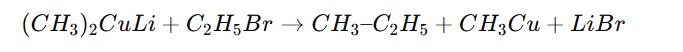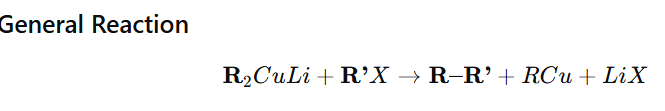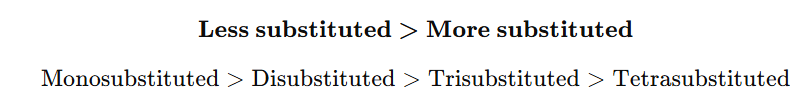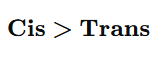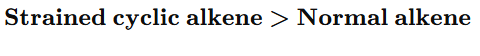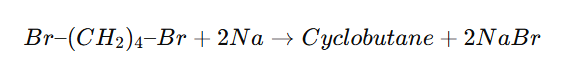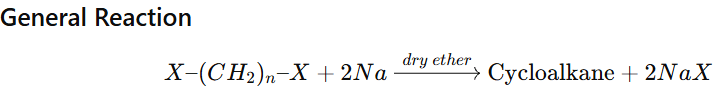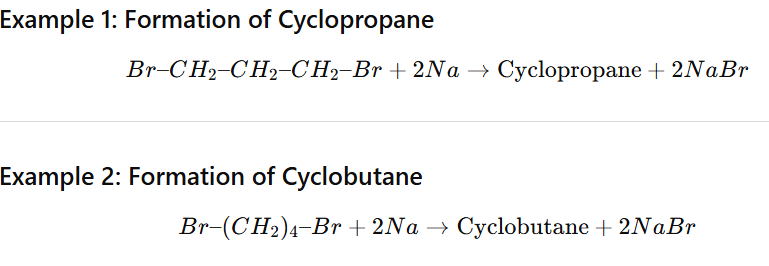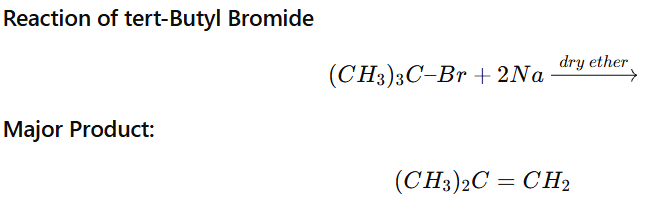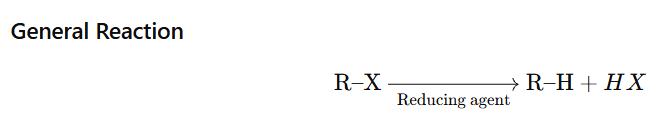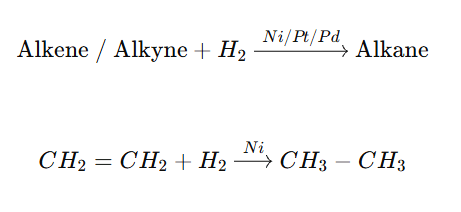Aromatisation is the conversion of higher alkanes (C₆ or more) into aromatic hydrocarbons (benzene and its derivatives) by dehydrogenation and cyclisation at high temperature in presence of catalysts.
n-Octane (C₈) → Xylene (or ethylbenzene)
Also possible under catalytic reforming conditions.
Theoretical Point of View
There is no fixed maximum carbon limit. Any C₆ or higher straight-chain alkane (C₆⁺) can undergo aromatisation because at least six carbons are required to form a benzene ring.
🔹 Practical / Industrial Point of View
In petroleum refining (catalytic reforming):
✔ Most effective for C₆ to C₁₀ alkanes ✔ Above C₁₀–C₁₂, cracking and side reactions increase ✔ Very long chains prefer cracking rather than clean aromatisation
So practically:
Cyclohexane undergoes multiple dehydrogenations:
👉 Total 4 molecules of H₂ released.
🔹 Key Concept
✔ First dehydrogenation, then cyclisation, then aromatic stabilization ✔ Metal catalyst helps in removal of hydrogen ✔ Final product gains extra stability due to aromatic resonance
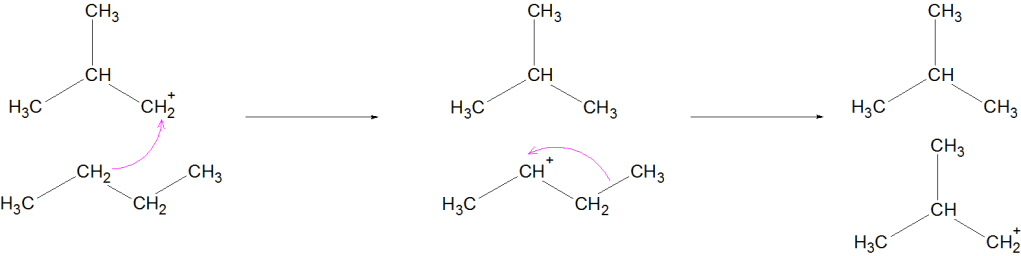
Isomerisation of alkane is the process in which a straight-chain alkane (n-alkane) is converted into its branched-chain isomer without changing the molecular formula.
Conditions Required
Catalyst:
Anhydrous AlCl₃
HF
Pt/Al₂O₃
Temperature: 250–400°C (industrial process)
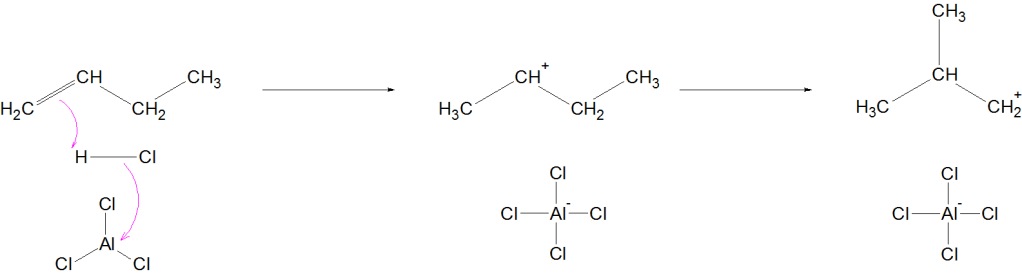
Occurs via carbocation mechanism
Mechanism (Simplified for JEE/NEET)
Formation of carbocation
Hydride shift or methyl shift
Formation of more stable branched carbocation
Deprotonation → Branched alkane
Industrial Importance
Used in petroleum refining
Improves octane number of petrol
Branched alkanes burn more smoothly (less knocking)
Corey–House synthesis is a method used to prepare higher alkanes by coupling an alkyl halide with a Gilman reagent (lithium dialkylcuprate, R₂CuLi).
Preparation of Gilman Reagent
Important JEE/NEET Points
✔ Best with primary alkyl halides ✔ Does not work well with tertiary halides (elimination dominates) ✔ Useful for forming unsymmetrical alkanes ✔ Milder and more selective than Wurtz reaction ✔ Works via substitution mechanism
“Hydrogenation rate of cyclic alkenes depends on ring strain. Smaller rings possess greater angle strain and hence higher heat of hydrogenation. Therefore, they react faster.”
JEE/NEET Important Order
Rate of hydrogenation:
Comparison Example (Very Important for Exams)
Compare:
1,3-Butadiene (conjugated)
1,4-Pentadiene (isolated)
Stability Order:
Rate of Hydrogenation:
JEE/NEET Important Order
Rate of hydrogenation:
(Benzene hydrogenates very slowly due to aromatic stability)
✔ Only alkali metals (Na, K, Li) give Wurtz-type coupling ✔ Sodium is preferred due to controlled reactivity ✔ Mg and Zn mainly form organometallic intermediates instead of direct alkane
Wurtz Reaction – Metals Other Than Sodium
Although sodium (Na) is the classical metal used in Wurtz reaction, some other metals can also promote coupling of alkyl halides
Potassium (K)
✔ More reactive than sodium ✔ Reaction is more vigorous ❌ Harder to control 📌 Rarely used in practice (safety issue)
Lithium (Li)
✔ Can participate in similar coupling ✔ Often forms organolithium intermediates 📌 Used more in organometallic synthesis than classical Wurtz
Silver (Ag)
✔ Used mainly for radical formation studies ❌ Not common for alkane synthesis 📌 Often used in rearrangement or carbocation studies
Zinc (Zn)
Zinc does not give classical Wurtz coupling easily but forms:
✔ Forms organozinc compounds ✔ Used in coupling reactions (e.g., Reformatsky type)
Magnesium (Mg)
R–X+Mg→R–MgX
✔ Forms Grignard reagent ❌ Does NOT directly give Wurtz alkane 📌 Very important alternative pathway
Cyclic Wurtz Reaction (Intramolecular Wurtz)
Cyclic Wurtz reaction is an intramolecular version of the Wurtz reaction in which a dihaloalkane reacts with sodium metal in dry ether to form a cycloalkane.
JEE / NEET Important Points
Best for small rings (3–5 members).
Large rings are difficult due to entropy factor.
Competes with intermolecular Wurtz (polymerization possible).
Primary dihalides give better yield.
Dry ether is essential.
Mechanism involves free radicals.
Product of Tertiary Alkyl Halide in Wurtz Reaction
When a tertiary alkyl halide (3° RX) is treated with sodium (Na) in dry ether, it does not give Wurtz coupling product efficiently.
Major product = Alkene (Elimination product) Coupling product is minor or negligible.
Example
Why Elimination Occurs?
✔ Tertiary halides easily form stable tertiary radicals / carbanions ✔ Strong base character of sodium promotes β-elimination ✔ Steric hindrance prevents effective coupling
Thus, E2-type elimination dominates over coupling.
JEE / NEET Important Points
Primary RX → best for Wurtz coupling
Secondary RX → mixture
Tertiary RX → alkene (major)
Elimination increases with substitution.
Reaction follows radical pathway but elimination competes strongly.
Reactivity Order (Bond Strength Basis)
JEE / NEET Important Points
Alkyl bromides are preferred.
Primary halides give best results.
Aryl halides do NOT undergo normal Wurtz (need Wurtz–Fittig).
Fluorides are practically inactive.
Dry ether is essential for the reaction.
Reasons for Using Dry Ether
1. Provides Anhydrous Medium
✔ Sodium reacts violently with water:
If moisture is present, sodium will react with water instead of alkyl halide. Therefore, ether must be dry.
Why is solvent needed in Wurtz reaction? Solvent used → Dry ether 1. Medium for Reaction Sodium is solid Alkyl halide is liquid Reaction occurs at sodium surface Ether provides: Proper contact between reactant.
2. Uniform reaction medium
Without solvent → reaction is uncontrolled / incomplete. 3. Stabilizes Reactive Intermediates Wurtz reaction proceeds via: Radical pathway or Organosodium intermediate (R–Na) Dry ether:
MCQ Questions for practice:
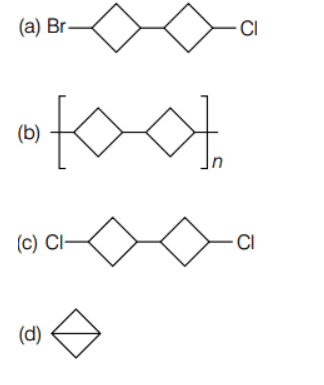
In the reaction of 1-bromo-3-chlorocyclobutane with two equivalents of sodium in ether, the major product is
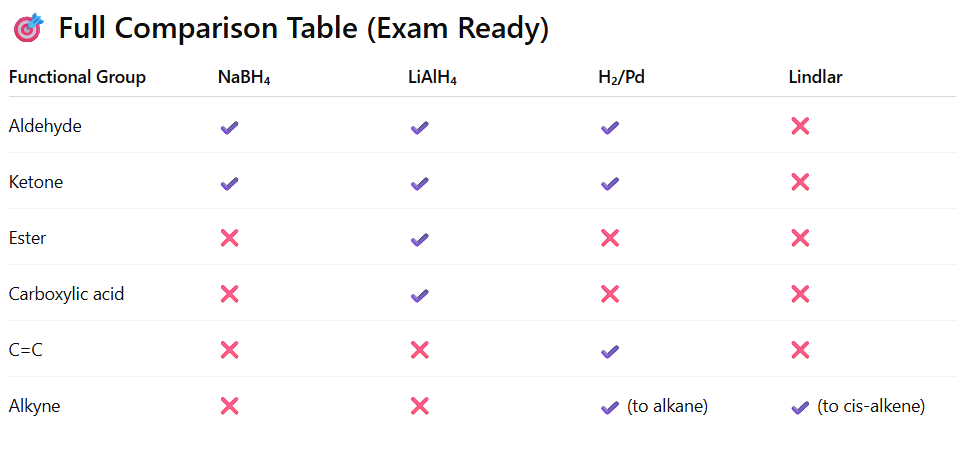
Alkyl halides can be reduced to alkanes by replacing the halogen atom with a hydrogen atom. This reduction typically involves the use of reducing agents.
Alkyl halides are reduced to alkanes by:
Nascent hydrogen (Zn/HCl)
Zn/CH3COOH
Zn + NaOH
LiAlH₄
Catalytic hydrogenation (H₂/Pd or Ni)
Zn- Cu Couple +C2H5OH
TPH (Triphenylhydride / Triphenyltin Hydride)
Via Grignard reagent followed by hydrolysis
Important Note for JEE/NEET
Reduction with Nascent Hydrogen Common in laboratory conditions.
Reduction with Hydrogen (Catalytic Hydrogenation)
Using H₂ with Pd/C, Ni or Pt catalyst , Effective especially for benzyl and allyl halides.
LiAlH₄ (Lithium aluminium hydride) Very effective for primary and secondary halides not for 3 degree.With LiAlH₄
Hydride (H⁻) acts as a strong nucleophile. Mechanism: Direct SN2 displacement of X⁻ by H⁻
NaBH4 and TPH are used to reduce 3 degree haloalkane to alkane.
Zn–Cu couple reduces alkyl halides to alkanes (especially in alcohol medium). Zn–Cu More Reactive than Zn? Copper coating removes oxide layer from zinc, increases surface activity , facilitates electron transfer, acts as better reducing system.
Reduction of Alkyl Halides by TPH. Reaction proceeds via free radical mechanism
The Sabatier–Senderens reaction is a catalytic hydrogenation reaction in which unsaturated compounds (alkenes or alkynes) are reduced to saturated compounds (alkanes) using hydrogen gas (H₂) in the presence of a metal catalyst such as Ni, Pt, or Pd.
It is named after the French chemists
Paul Sabatier
Jean-Baptiste Senderens
Paul Sabatier received the 1912 Nobel Prize in Chemistry for his work on catalytic hydrogenation.
What JEE / NEET Can Ask
Concept-Based Questions
Type of catalysis → Heterogeneous catalysis
Catalyst used → Ni (most common), Pt, Pd
Nature of reaction → Addition (Reduction) reaction
Mechanism → Adsorption theory
Mechanism (Very Important for JEE)
✔ Hydrogen adsorbs on Ni surface ✔ H–H bond breaks → Atomic hydrogen formed ✔ Alkene/Alkyne adsorbs ✔ Syn addition of hydrogen ✔ Alkane formed
🔥 Very Important Concept
✔ Syn addition may give cis product ✔ But syn ≠ cis always
Example:
Hydrogenation of cycloalkene → gives cis product But hydrogenation of 2-butene → gives alkane (no cis/trans left)
So in that case: Syn addition happened But cis concept disappears
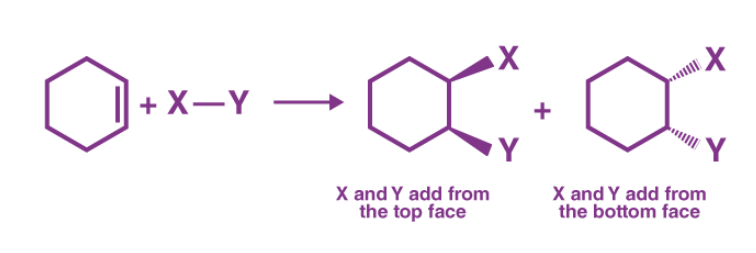
Syn Addition (Mechanism term)
Refers to how groups are added during a reaction.
Both substituents add from the same face of the double bond.
It is a mechanistic term.
Example: Hydrogenation using H₂/Ni (Sabatier–Senderens) → Syn addition
What is Syn Addition?
The addition of two substituents to the same side (or face) of a double or triple bond reduces the bond order but increases the number of substituents.
Raney nickel is significantly more reactive than standard nickel powder. It is Created from a
Nickel-Aluminum alloy, where Aluminum is removed by caustic leaching (NaOH)
Platinum (like Ni, Pd) mainly prefers:
In a benzylidene compound containing a ketonic (C=O) group, when is only the C=C reduced?
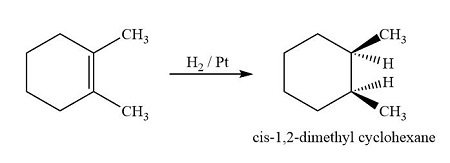
NOTE : Pd isomerises the alkene hence with Pd , trans -isomer predominates.
Palladium can catalyze alkene isomerisation via reversible adsorption on its surface, leading to thermodynamic control where the more stable trans isomer predominates.
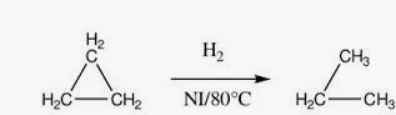
Note: When Cyclopropane/butane reacts with hydrogen gas in the presence of Ni, it gives propane/butane
he ring opens due to high strain and behave like an alkene
Cyclobutane also has ring strain (90° vs 109.5°)
Less than cyclopropane but still significant
Ring opens under catalytic conditions
Important JEE/NEET Concept
Reactivity order due to ring strain: Cyclopropane> Cyclobutane > Cyclopentane
Cyclopentane and cyclohexane usually do NOT open easily.