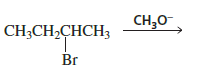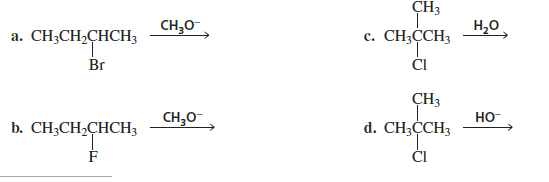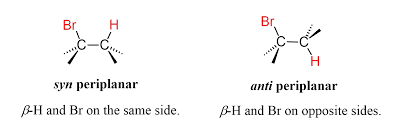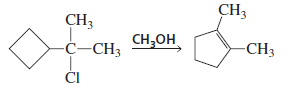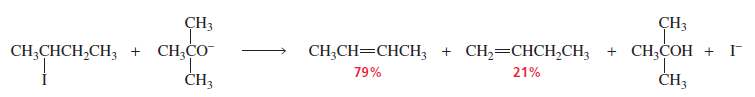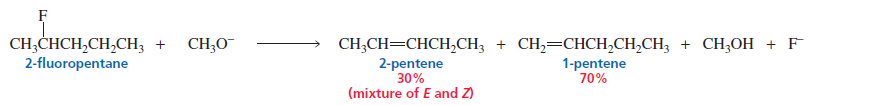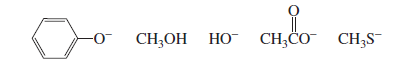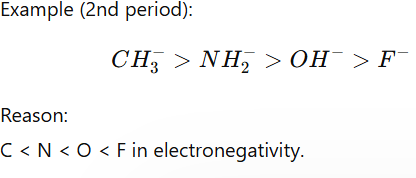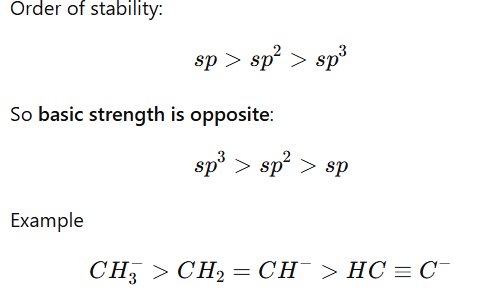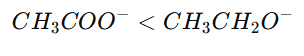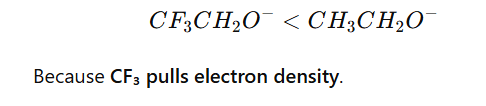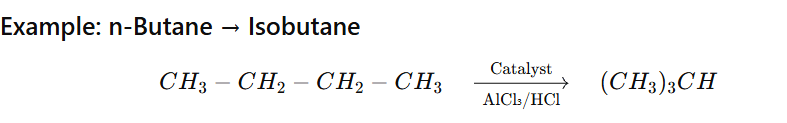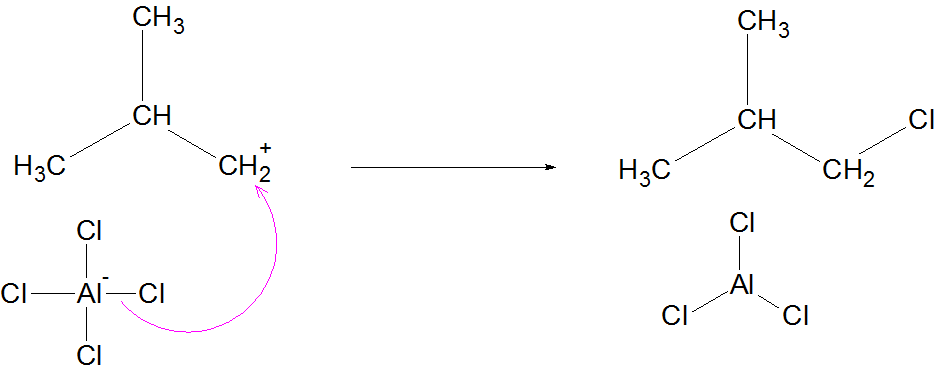Sarvan Kumarhttp://sarvankumar.wordpress.comMy name is Sarvan Kumar, I am post graduate in chemistry (MSc). besides teaching chemistry at various schools and coaching centres I have been giving home tuitions to students for 10 years. I have helped students score good marks in chemistry not only in board examination but also JEE, NEET, SAT, IGCSE and IB examinations. Nearly 90% of my students have scored more than 95% in their CBSE board examination. Moreover they have also secured a seat in prestigious engineering and medical college.
Since every student is different hence my unique style of teach
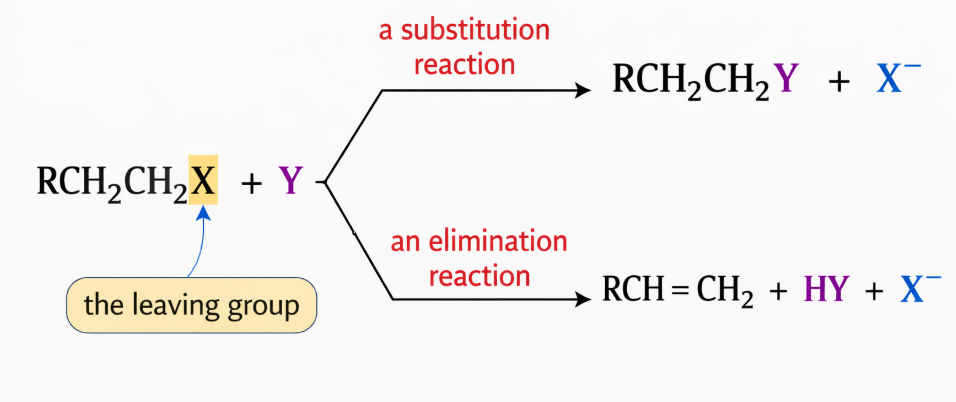
In organic chemistry, competition between E1 Reaction and E2 Reaction occurs when a substrate and reaction conditions allow both elimination pathways. The dominant mechanism depends on several factors.
Primary and secondary alkyl halides undergo elimination mainly through the E2 mechanism because the carbocations that would form in an E1 Reaction are relatively unstable, and solvolysis reactions usually involve a high concentration of base that favors the E2 Reaction.
However, tertiary alkyl halides can undergo both E2 and E1 reactions because they are capable of forming relatively stable tertiary carbocations.
Because tertiary alkyl halides are able to undergo both E2 and E1 elimination reactions, the overall rate of the reaction depends on both pathways. Therefore, the rate law is the sum of the rate laws for the E2 and E1 reactions.
Thus, An E2 reaction is favored by a high concentration of a strong base. An E1 reaction is favored by a low concentration of a weak base
For each of the following reactions, (1) decide whether an E2 or an E1 occurs, and (2) draw the major elimination product:
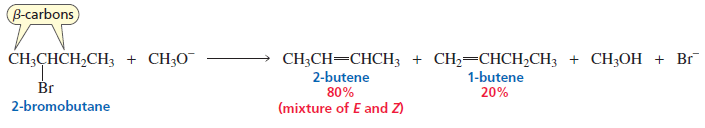
When 2‑Bromobutane reacts with Methoxide Ion, the reaction mainly proceeds through an E2 Reaction because:
2-Bromobutane is a secondary alkyl halide.
CH₃O⁻ (methoxide) is a strong base.
Strong bases favor E2 elimination.
The elimination forms bu-2-tene:
Major product: 2‑Butene (especially trans-2-butene, most stable)
Minor product: 1‑Butene
Reason (Zaitsev rule)
According to Zaitsev Rule, the more substituted alkene forms as the major product.
Order of products:
Substrate: secondary alkyl halide
Base: strong (CH₃O⁻)
Mechanism: E2 elimination
Major product: trans-2-butene
Problem: For each of the following reactions, (1) decide whether an E2 or an E1 occurs, and (2) draw the major elimination product:
Strong base → E2
Weak base + tertiary substrate → E1
Zaitsev alkene = major product
let’s analyze each reaction using two steps: Decide E1 or E2 Write the major elimination product
(a)
Substrate: 2-Bromobutane Reagent: CH₃O⁻ (methoxide) → strong base
Reasoning
Secondary alkyl halide
Strong base present E2 mechanism dominates
Elimination (Zaitsev rule) The base removes a β-hydrogen giving the more substituted alkene.
Ring expansion is a type of carbocation rearrangement in which a smaller ring increases its size (usually by one carbon) to reduce ring strain.
It commonly occurs in SN1 or E1 reactions when a carbocation is adjacent to a small ring.
Why Ring Expansion Happens
Small rings such as:
Cyclopropane
Cyclobutane
have high angle strain.
When a carbocation forms next to these rings, a C–C bond shifts (1,2-shift) and the ring expands to:
Cyclobutane
Cyclopentane
which are more stable
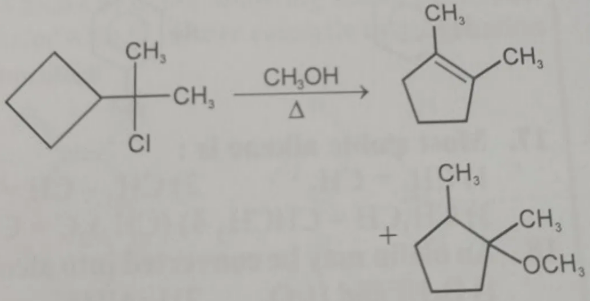
PROBLEM : Propose a mechanism for the following reaction:
In this reaction the substrate is a tertiary alkyl chloride attached to a cyclobutane ring and the reagent is methanol (CH₃OH), which is a weak nucleophile and polar protic solvent.
So the reaction proceeds through an SN1 mechanism.
Step 1: Carbocation formation
The Cl⁻ leaves, forming a tertiary carbocation.
Step 2: Ring expansion (rearrangement)
Cyclobutane rings have high ring strain. To reduce strain, a 1,2-shift of a ring bond occurs, converting:
cyclobutane → cyclopentane carbocation
This produces a more stable carbocation and also reduces ring strain.
Step 3: Deprotonation
Methanol removes a proton from the carbocation, giving an alkene.
Final Product
The rearranged product is:
1,2-dimethylcyclopentene
In this reaction substitution does NOT take place as the final product
Reason
Leaving group departure Cl⁻ leaves first → tertiary carbocation forms (SN1 condition, because solvent is CH₃OH, weak nucleophile).
Carbocation rearrangement The carbocation is next to a cyclobutane ring, which has high ring strain. So a 1,2-C–C shift occurs, causing ring expansion:
Cyclobutane → Cyclopentyl carbocation
This rearrangement is very favorable because it relieves ring strain.
Elimination occurs Instead of nucleophilic attack by methanol, a proton is removed, forming a stable alkene.
Why elimination dominates
Rearranged carbocation becomes very stable
Formation of cyclopentene double bond is thermodynamically favorable
Relief of ring strain drives the reaction
Final product
1,2-dimethylcyclopentene
✔ Therefore substitution product (ether) is not observed as the major product. The reaction proceeds via SN1 → rearrangement → E1 elimination.
Important teaching point (useful for students)
Whenever you see:
Carbocation next to small ring (cyclobutane / cyclopropane)
Ring expansion possible
➡ Rearranged alkene products are usually formed.
If you want, I can also show the step-by-step rearrangement mechanism diagram (very useful for explaining this in class).
Can cyclopentane undergo ring expansion?
Yes, but it is much less common.
Reason
Ring expansion mainly occurs to reduce ring strain.
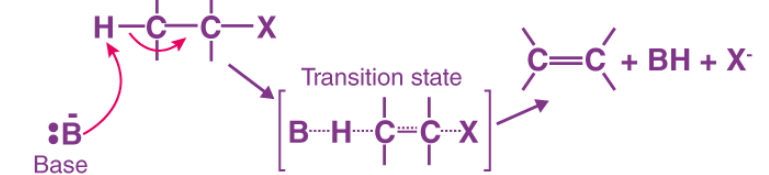
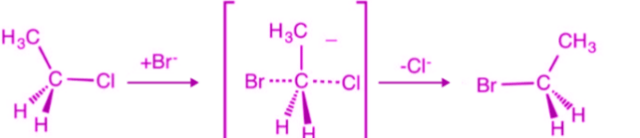
A base removes a proton (H⁺) from a carbon atom that is adjacent to the carbon bonded to the halogen. As the proton is removed, the electrons from the C–H bond move toward the neighboring carbon that is attached to the halogen. These electrons then form a double bond between the two carbons. At the same time, the halogen leaves with its bonding electron pair, because carbon cannot form more than four bonds. This process results in the formation of an alkene.
Why E2 Reaction is Regioselective
An E2 reaction is called regioselective because the elimination of hydrogen and the leaving group can produce more than one possible alkene, but one alkene is formed in greater amount than the others.
Reason
In E2 elimination, the base removes a β-hydrogen (hydrogen on the carbon adjacent to the carbon bearing the leaving group). If there are different β-carbons, the base can remove hydrogen from different positions, giving different alkenes.
However, according to Zaitsev’s rule, the more substituted alkene (the alkene with more alkyl groups attached to the double-bonded carbons) is usually more stable and therefore forms as the major product.
Zaitsev’s Rule
Alexander M. Zaitsev, a nineteenth-century Russian chemist, proposed a rule to predict the major alkene formed in elimination reactions.
According to Zaitsev’s rule, the major product is the more substituted alkene. This occurs when the base removes a hydrogen from the β-carbon that has fewer hydrogen atoms.
As a result, the double bond forms between the more substituted carbon atoms, producing the more stable alkene.
Limitations of Zaitsev’s Rule (JEE / NEET level)
Although Zaitsev’s rule usually predicts the major alkene in elimination reactions, there are several situations where it does not apply.
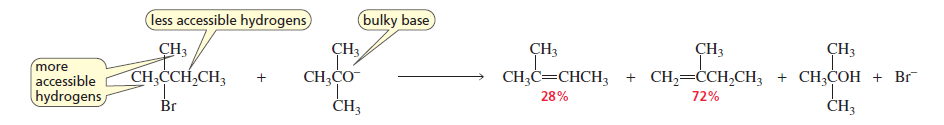
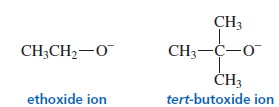
1. Bulky (Sterically Hindered) Bases
When a bulky base is used, it removes the most accessible hydrogen instead of the one predicted by Zaitsev’s rule.
Examples of bulky bases:
tert-butoxide (t-BuO⁻)
LDA
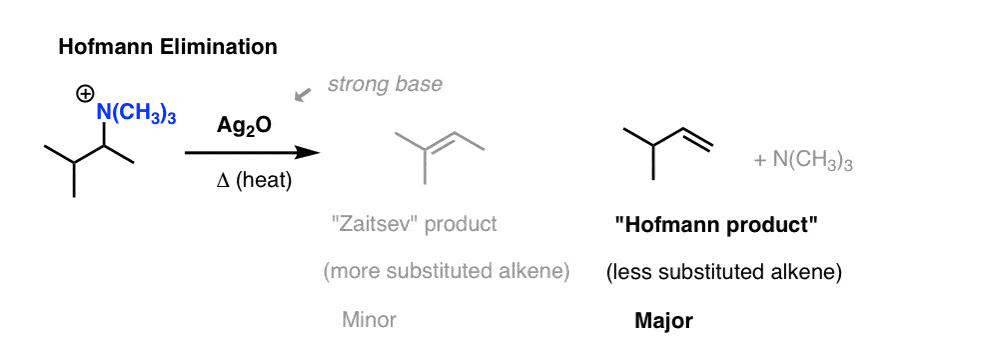
Result → Less substituted alkene (Hofmann product) becomes the major product.
If the alkyl halide is not sterically hindered and the base is only moderately hindered, the more stable alkene will be the major product, as expected. In other words, it takes a lot of steric hindrance for the less stable product to be the major product. Thus, the major product of the following reaction is 2-butene.
2. Poor Leaving Groups (e.g., Fluoride)
When the leaving group is F⁻, elimination often gives the less substituted alkene instead of the Zaitsev product.
Reason → Strong C–F bond and different transition state stability.
When a hydrogen and a chlorine, bromine, or iodine are eliminated from an alkyl halide, the halogen starts to leave as soon as the base begins to remove the proton. Consequently, the transition state resembles an alkene (see page 414). The fluoride ion, however, is the strongest base of the halide ions and, therefore, the poorest leaving group.
Elimination of quaternary ammonium hydroxides produces mainly the less substituted alkene.
The Hofmann elimination is the preparation of alkenes from the treatment of quaternary ammonium salts with silver oxide, water, and heat.
General features: The Hofmann elimination is a β-elimination. Thus, the β-hydrogen is abstracted by the base (hydroxide ion) from the β-carbon atom. It is also an anti elimination (the leaving group has to be antiperiplanar). Besides, some side reactions occur when the base acts as a nucleophile, delivering then alcohol byproducts.
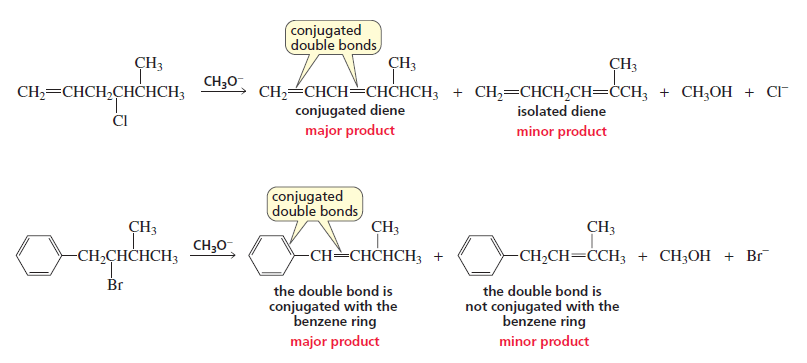
4. In each of the following reactions, the major product is the alkene with conjugated double bonds because it is the more stable alkene, even though it is not the more substituted alkene.
Relative Reactivities in an E2 Reaction
In E2 elimination, the rate depends on how easily the base can remove a β-hydrogen and how stable the resulting alkene will be.
Elimination from more substituted alkyl halides usually forms more substituted and more stable alkenes. Therefore, the reaction occurs more easily.
Reactivity Order of Alkyl Halides in E2
Explanation
Tertiary alkyl halides (3°) eliminate fastest because they form highly substituted stable alkenes.
Secondary alkyl halides (2°) react at a moderate rate.
Primary alkyl halides (1°) react slowly because they form less substituted alkenes.
Methyl halides (CH₃X)do not undergo E2 because there is no β-hydrogen available for elimination.
Features of E2 Reaction (JEE / NEET level)
Bimolecular Reaction
Two species participate: alkyl halide and base.
Second-Order Kinetics
One-Step (Concerted) Mechanism
Proton removal, double bond formation, and leaving group departure occur simultaneously.
Strong Base Required
Examples: OH⁻, RO⁻, t-BuO⁻, NH₂⁻
β-Elimination Reaction
Hydrogen is removed from the β-carbon (adjacent to the carbon bearing the halogen).
No Carbocation Intermediate
Reaction occurs in one step, so rearrangement does not occur.
Anti-Periplanar Requirement
The β-hydrogen and leaving group must be in opposite (anti) orientation.
Regioselective Reaction
Usually follows Zaitsev’s rule → more substituted alkene forms as major product.
Effect of Substrate Structure Reactivity order:
3∘>2∘>1∘
Competes with SN2 Reaction
Strong bases with secondary or tertiary halides often favor E2 elimination.
Stereochemistry of E2 Mechanism
The E2 (Elimination Bimolecular) reaction has very important stereochemical requirements. For elimination to occur, the β-hydrogen and leaving group must have a specific spatial arrangement.
1. Anti-Periplanar Requirement
The β-hydrogen (H) and leaving group (X) must lie in the same plane but opposite directions (180° apart).
This arrangement is called anti-periplanar geometry.
Reason: This alignment allows proper overlap of orbitals so the π bond of the alkene can form easily.
2. Concerted Mechanism
E2 occurs in one single step:
Base removes β-H
C–H bond breaks
C–X bond breaks
C=C double bond forms simultaneously
There is no intermediate (no carbocation).
3. Stereospecific Reaction
Because the geometry must be anti-periplanar, the reaction is stereospecific.
Example:
Different stereoisomers of reactants can give different alkene stereoisomers (E or Z).
4. Anti Elimination
Most E2 reactions proceed by anti elimination because:
It is energetically more stable
Less steric repulsion between groups.
5. Syn Elimination (Rare)
If anti arrangement is not possible, syn-periplanar elimination may occur.
However, it is rare and higher in energy.
6. Important in Cyclic Compounds
In cyclohexane systems:
Leaving group must be axial
β-Hydrogen must also be axial This creates the required anti-periplanar arrangement.
7. Product Stereochemistry
E2 generally favors formation of the more stable alkene (Zaitsev product), though bulky bases may give Hofmann product.
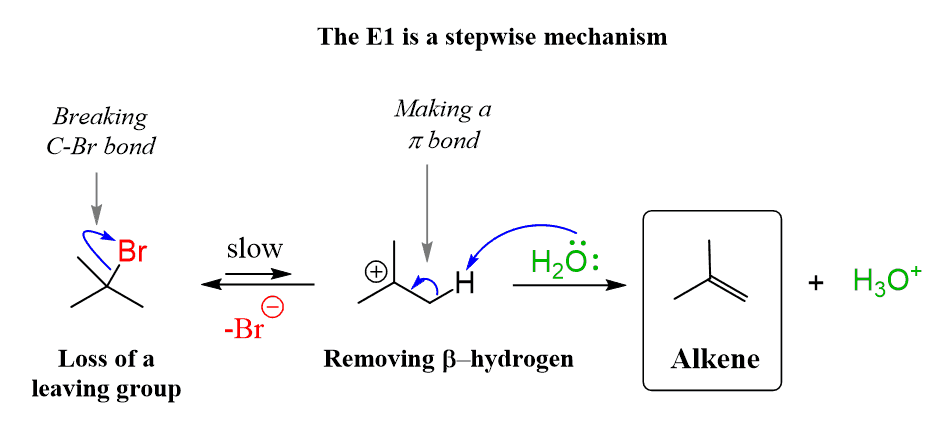
THE E1 REACTION
E1 Mechanism (Elimination Unimolecular) is a two-step elimination reaction commonly seen with tertiary alkyl halides in polar protic solvents. These are the main features important for JEE/NEET:
1. First-Order Kinetics
Rate depends only on the concentration of alkyl halide.
Rate law:
Rate=k[R–X]
2. Two-Step Mechanism
Slow step: Leaving group leaves → carbocation formation
Fast step: Base removes β-hydrogen → alkene formation
3. Carbocation Intermediate
A planar carbocation is formed.
Because of this, rearrangement (hydride shift or methyl shift) can occur.
4. Favored with Tertiary Alkyl Halides
Carbocation stability determines reactivity:
3° > 2° >> 1°
Primary carbocations are unstable, so E1 rarely occurs with primary halides.
5. Polar Protic Solvent Favours E1
Solvents like:
H₂O
Alcohols (ROH)
These stabilize the carbocation and leaving group.
6. Weak Bases Are Sufficient
Strong base is not required because the slow step is carbocation formation.
7. Zaitsev Alkene Major Product
The more substituted (more stable) alkene is usually the major product.
8. Competes with SN1 Reaction
Because both involve carbocation intermediate, E1 and SN1 often occur together.
9. Rearrangement Possible
Carbocation may rearrange to form a more stable carbocation, giving unexpected products.
10. Usually Occurs at Higher Temperature
Higher temperature favors elimination over substitution.
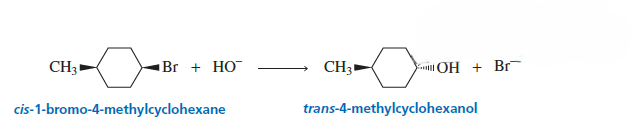
both E1 and E2 reactions can give cis and trans alkenes, but the reason and control are different.
In E1 Reaction
Carbocation intermediate forms (planar).
Base can remove β-H from either side.
Therefore both cis (Z) and trans (E) alkenes form.
Trans usually major because it is more stable.
Not stereospecific.
In E2 Reaction
Reaction occurs in one step.
Requires anti-periplanar geometry between β-H and leaving group.
The geometry of the reactant determines the alkene formed.
So cis or trans may form depending on the starting stereochemistry.
Stereospecific reaction.
Stereochemistry of E1 Mechanism
The E1 (Elimination Unimolecular) reaction has different stereochemistry from E2 because it proceeds through a carbocation intermediate.
1. Planar Carbocation Intermediate
In the first step, the leaving group (X) leaves and forms a carbocation.
The carbocation is sp² hybridized and planar.
Because it is planar, the β-hydrogen can be removed from either side.
2. Not Stereospecific
Unlike E2, E1 is not stereospecific.
Reason: Since the intermediate carbocation is planar, the base can remove the β-hydrogen from different orientations, giving different alkene stereoisomers.
3. Formation of E and Z Alkenes
Both E (trans) and Z (cis) alkenes can form.
However:
Trans (E) alkene is usually the major product
because it is more stable (less steric repulsion).
4. No Anti-Periplanar Requirement
In E1, the β-hydrogen and leaving group do not need anti-periplanar geometry.
This is because:
the leaving group already left in the first step.
5. Rearrangement Possible
Since a carbocation intermediate exists, rearrangements such as:
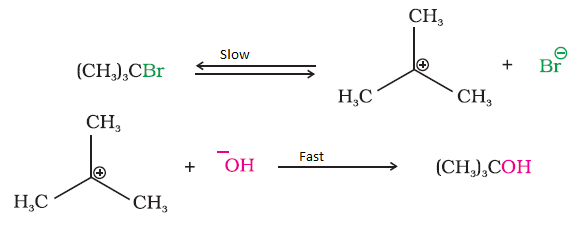
Step 1: Formation of carbocation (slow, rate-determining)
Step 2: Nucleophile attack (fast)
Carbocation Intermediate Forms
Stability of carbocation controls reaction rate.
Reactivity Order of Substrate
Polar Protic Solvents Favor SN1
Examples: H₂O, ROH, ethanol
Weak Nucleophile is Sufficient
Examples: H₂O, ROH
Carbocation Rearrangement Possible
Hydride shift or methyl shift may occur.
Racemization of Product
If the carbon is chiral, product becomes racemic mixture.
Good Leaving Group Required
Most SN1 reactions give partial racemization. In an SN1 reaction, a carbocation intermediate is formed, which is planar. Because of this planar structure, the nucleophile can attack from both sides, leading to formation of two stereoisomers.
However, in most cases the products are not formed in equal amounts. Usually, 50–70% of the product has inverted configuration, while the rest has retained configuration.
If equal amounts of both stereoisomers are produced, the reaction is called complete racemization. When more of the inverted product is formed, the reaction is called partial racemization
Factors Affecting SN1 Reactions
The rate of an SN1 reaction is influenced mainly by the leaving group and the nucleophile.
Effect of Leaving Group in SN1 Reaction
In an SN1 reaction, the rate-determining step is the formation of a carbocation. Therefore, the reaction rate depends on two important factors:
Ease of leaving group departure
The reaction is faster when the leaving group can easily dissociate from the substrate.
Stability of the carbocation formed
The more stable the carbocation, the faster the SN1 reaction occurs.
Therefore, good leaving groups and stable carbocations increase the rate of SN1 reactions.
PROBLEM : Which of the following reactions take place more rapidly when the concentration of the nucleophile is increased?
he question asks: Which reaction becomes faster when the nucleophile concentration increases?
Key concept:
SN2 reaction: Rate = k[R–X][Nu−] → depends on nucleophile concentration
SN1 reaction: Rate = k[R–X] → independent of nucleophile concentration
So we must identify which reactions proceed by SN2.
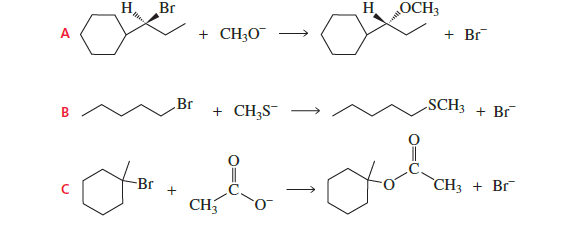
Reaction A
Substrate: secondary alkyl bromide attached to cyclohexane
This structure favors SN1 (carbocation formation possible).
Therefore rate does NOT depend strongly on nucleophile concentration.
❌ Not the answer
Reaction B
Substrate: primary alkyl bromide
Primary halides undergo SN2 easily.
Rate depends on nucleophile concentration (CH₃S⁻).
Nucleophilicity means the ability of a species to donate an electron pair to an electrophile and form a bond. In exams, students must quickly compare nucleophiles using a few core rules.
When we talk about atoms or molecules that have lone-pair electrons, sometimes we call them bases and sometimes we call them nucleophiles (Table 9.1). What is the difference between a base and a nucleophile?
Basicity tells us how easily a compound (base) can donate its lone pair of electrons to a proton (H⁺). A strong base donates its electron pair more easily than a weak base.
Basicity is related to the acid dissociation constant (Ka) of its conjugate acid, which shows how easily that conjugate acid releases a proton. If the conjugate acid releases H⁺ easily, the base is weaker; if it does not release H⁺ easily, the base is stronger.
Nucleophilicity describes how easily a compound (nucleophile) can attack an electron-deficient atom and form a new bond. It is measured by the rate constant (k), which indicates how fast the nucleophile reacts in a chemical reaction.
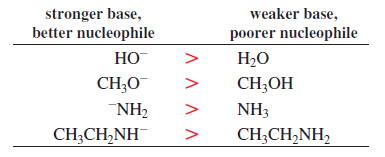
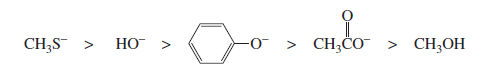
Note: Species with a negative charge is a stronger base and a better nucleophile than a species that has the same attacking atom but is neutral
If the attacking atoms are the same size, stronger bases are better nucleophiles.
−NH2 > HO− > F−
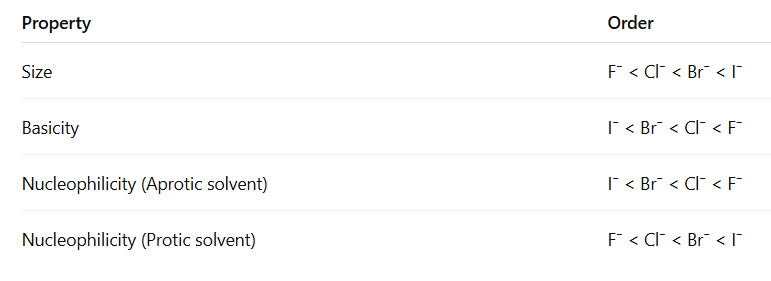
If, however, the attacking atoms of the nucleophiles are very different in size, another factor comes into play: the polarizability
Effect of Solvent on Nucleophilicity (Simple Point-wise) 🧪
Species with negative charge are stronger bases than neutral ones.
Example
Basicity is a measure of how well a compound (a base) shares its lone pair with a proton. The stronger the base, the better it shares its electrons. Basicity is measured by an equilibrium constant (the acid dissociation constant, Ka) that indicates the tendency of the conjugate acid of the base to lose a proton
2. Electronegativity (Across a Period)
If atoms are in the same period, basicity decreases with electronegativity.
Less electronegative atom holds negative charge poorly → stronger base.
3.Hybridization Effect
More s-character stabilizes negative charge, making the base weaker.
4.Resonance
If negative charge is delocalized, the base becomes less basic.
Example:
Acetate ion vs Ethoxide
5.Inductive Effect
Electron-withdrawing groups (–I effect) decrease basicity.
Example
6. Aromaticity
If negative charge contributes to aromatic stabilization, the base becomes weaker.
Example Cyclopentadienyl anion is stable → weak base.
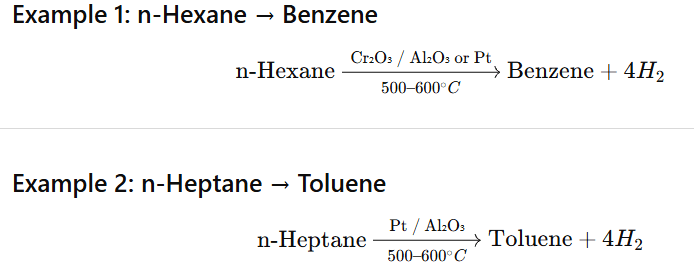
Aromatisation is the conversion of higher alkanes (C₆ or more) into aromatic hydrocarbons (benzene and its derivatives) by dehydrogenation and cyclisation at high temperature in presence of catalysts.
n-Octane (C₈) → Xylene (or ethylbenzene)
Also possible under catalytic reforming conditions.
Theoretical Point of View
There is no fixed maximum carbon limit. Any C₆ or higher straight-chain alkane (C₆⁺) can undergo aromatisation because at least six carbons are required to form a benzene ring.
🔹 Practical / Industrial Point of View
In petroleum refining (catalytic reforming):
✔ Most effective for C₆ to C₁₀ alkanes ✔ Above C₁₀–C₁₂, cracking and side reactions increase ✔ Very long chains prefer cracking rather than clean aromatisation
So practically:
Cyclohexane undergoes multiple dehydrogenations:
👉 Total 4 molecules of H₂ released.
🔹 Key Concept
✔ First dehydrogenation, then cyclisation, then aromatic stabilization ✔ Metal catalyst helps in removal of hydrogen ✔ Final product gains extra stability due to aromatic resonance
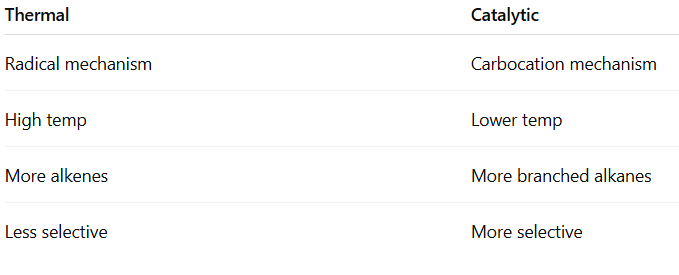
Isomerisation of alkane is the process in which a straight-chain alkane (n-alkane) is converted into its branched-chain isomer without changing the molecular formula.
Conditions Required
Catalyst:
Anhydrous AlCl₃
HF
Pt/Al₂O₃
Temperature: 250–400°C (industrial process)
Occurs via carbocation mechanism
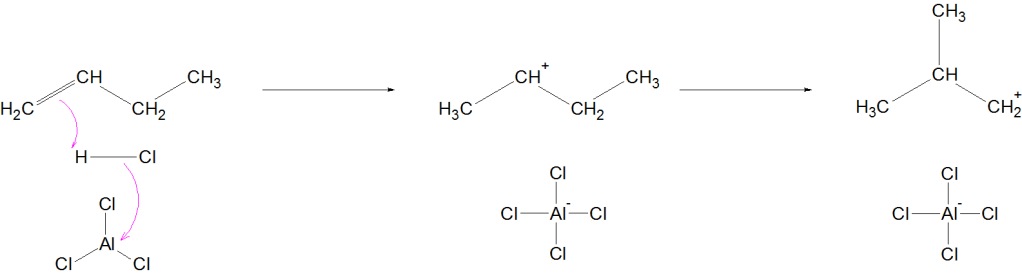
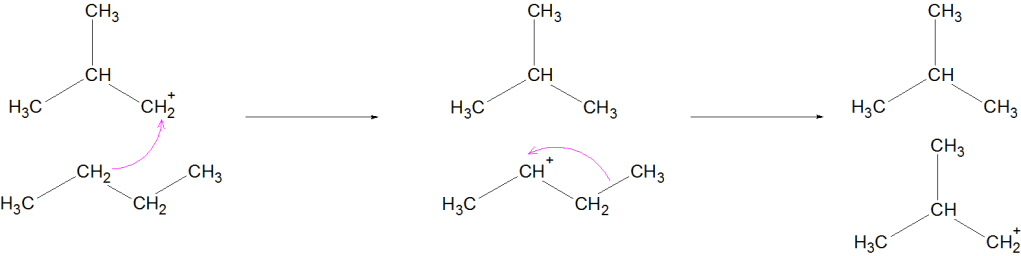
Mechanism (Simplified for JEE/NEET)
Formation of carbocation
Hydride shift or methyl shift
Formation of more stable branched carbocation
Deprotonation → Branched alkane
Industrial Importance
Used in petroleum refining
Improves octane number of petrol
Branched alkanes burn more smoothly (less knocking)